
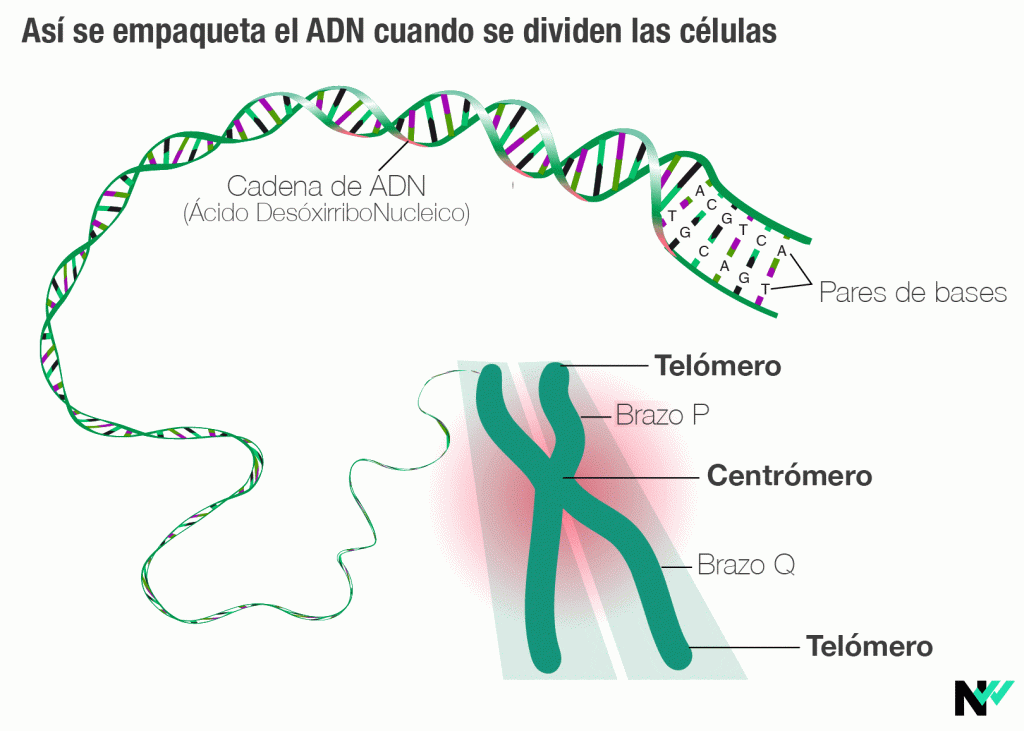
Nuestro libro de instrucciones genético se conserva en una cambiante biblioteca de ADN, empaquetada fundamentalmente en el núcleo de nuestras células. Es el genoma humano. Cuando se van a multiplicar, esos ‘libros’ de genes se organizan 36 grandes estanterías. Son los cromosomas. Esas estanterías se arman por pares de dos en dos cromosomas –salvo las células reproductivas–. Cromosomas XX, los femeninos; XY, los masculinos.
La cuestión es que encontrar un libro concreto puede volverse una odisea si ese libro (gen) se oculta en una parte de la estantería que coincide con una esquina. O bien, si el libro está justo al final del todo del pasillo, apretado en el extremo del lineal. En la vida real de nuestras células, eso sería buscar genes y bases (las letras del código ADN) en los centrómeros (parte central, donde se unen los ‘brazos’ del cromosoma) y los telómeros (extremos de los ‘brazos’ del cromosoma).
La biotecnóloga Karen Miga ha conseguido llegar a esos lugares de manera más rápida y eficiente. Esta investigadora de la Univiersidad de California Santa Cruz (EE.UU.) pertenece al consorcio ‘T2T’ (Telómero a Telómero), que acaba de publicar en Science la última y más completa versión del genoma humano.
“Hasta ahora hemos estado ciegos”, explica a Newtral.es desde California esta bióloga, que ha vivido una semana frenética. No todos los días se firma una portada en la revista científica que hace 21 años alumbró la primera versión de ese genoma humano incompleto. Sin embargo, es la segunda vez que descorcha champán en menos de un año. En mayo ya publicó la secuencia más completa del genoma humano en un repositorio abierto de ‘preprints’.
Todo esto está muy bien para el avance de la ciencia. Pero “servirá para mejorar la vida de la gente”. Aunque habrá que esperar. Por lo pronto, hoy han logrando completar un puzle complicado, lleno de piezas casi idénticas, repetitivas, que creíamos irrelevantes. “Este momento dará forma a décadas de descubrimientos e impulsarán tecnologías con una visión global”.
- PREGUNTA: ¿Por qué los centrómeros y los telómeros son tan complicados y qué información relevante (por ejemplo, sobre enfermedades) contienen?
- RESPUESTA: Se sabía que las brechas más grandes y persistentes en nuestro genoma se ubicaban en lugares que eran críticos para la vida. Por ejemplo, los centrómeros son las regiones importantes para garantizar que nuestros cromosomas se segreguen correctamente cada vez que una célula se divide. Si hay errores, puede conducir a cánceres y defectos de nacimiento. Esta es la primera vez que podemos estudiar las secuencias en estas importantes regiones. Además, no teníamos una lista completa de genes en nuestro genoma antes de nuestro trabajo, por lo que hemos avanzado nuestro catálogo para que sea más amplio y completo.
- P: ¿Cuándo cree que se podría utilizar esta versión como «referencia» del genoma humano en hospitales y centros de investigación?
- R: Aún no pueden usarlo, tendrá que pasar un tiempo antes de que los hospitales y profesionales de la medicina lo adopten por completo. En el futuro, cuando se secuencie el genoma de una persona, investigadores y médicos podrán estudiar su ADN de forma completa y más exhaustiva y, con suerte, utilizar esa información para guiar mejor su atención médica.
- P: ¿Y desde el punto de vista no estrictamente clínico?
- R: Conocer la secuencia completa del genoma humano proporcionará un marco integral para que los científicos estudien la variación, la enfermedad y la evolución del genoma humano.
Un ejemplo. El Hospital Universitario Gregorio Marañón de Madrid acaba de incorporar un equipo nuevo que permite secuenciar el genoma humano de una persona al completo. Una máquina con aspecto de fotocopiadora que puede comparar el resultado del paciente con los ‘moldes’ de genoma de referencia publicados desde 2001 (en concreto, la versión de 2013). Este hecho supone un avance a la hora de diagnosticar enfermedades.
A partir de los resultados, se abre un camino para personalizar tratamientos. Con suerte, se podrá estudiar si hay fármacos que pueden resultar efectivos en un paciente. Tecnologías que nacen el laboratorios y departamentos de ingeniería.
Para Miga ocurre algo así como que “lo mejor está por venir”. Entramos en los modelos científicos “del terreno de los sueños”, ya que no hay dos genomas humanos idénticos. Cada persona es genéticamente única. Es verdad que compartimos el 99,9% de esos ‘libros’ de genes. Pero lo interesante a veces se oculta en ese 0,1%. Una vez construido “el sueño”, llegarán “herramientas de mapeo y análisis de secuencias” que conviertan a este avance en algo particularmente útil para cada persona.
- P: La nueva técnica que han usado, de nanoporos, aunque más rápida, ¿no podría cometer más errores?
- R: Nuestro consorcio usó un enfoque multiplataforma, construyó ensamblajes repetidos inicialmente a partir de datos de alta fidelidad de PacBio, y también usó las lecturas extremadamente largas de Oxford Nanopore. Fue el uso de ambas tecnologías lo que permitió a nuestro equipo lanzar el ensamblaje terminado.
- P: ¿Cuáles son los retos pendientes? Se ha secuenciado sólo el cromosoma X, ¿nos perdemos algo importante con el Y?
- R: El genoma de referencia actual tampoco incluye un cromosoma Y (la línea celular contiene dos cromosomas X, es decir, se sacó de una mujer). Utilizamos un tipo de muestra diferente para completar este último cromosoma restante. El cromosoma Y es increíblemente repetitivo, con duplicaciones invertidas, genes amplicónicos [centrados en los testículos] y aproximadamente la mitad del cromosoma organizado en millones de bases de repeticiones en tándem.
- P: ¿Podemos decir que no hay un ‘genoma humano’ sino múltiples genomas humanos?
Es importante destacar que CHM13 (la actual versión) no captura la diversidad completa de la variación genética humana. Para abordar este sesgo, el Consorcio de Referencia del Pangenoma Humano se ha unido al Consorcio T2T para crear una colección de genomas de referencia de alta calidad de diversas poblaciones. Este será un enfoque crítico en los próximos años.
Como contaba a Newtral.es el biotecnólogo del CNB-CSIC Lluis Montoliu, “lo que quizá en un grupo de población sería considerado una mutación, en otro puede ser una variación poblacional y no va a tener ese significado patológico”. Por eso es importante que “si vamos a diagnosticar una enfermedad rara, si trabajamos con pacientes con familias españolas, es importante que tengamos un genoma de referencia que recoja la variabilidad intrínseca que hay en la población española”.



